Parte 1: Ejecutar un pipeline de demostración¶
Traducción asistida por IA - más información y sugerencias
En esta primera parte del curso de capacitación Hello nf-core, le mostramos cómo encontrar y probar un pipeline de nf-core, configurar y personalizar su ejecución según sus necesidades, y entender cómo la validación de entrada protege contra errores comunes.
Vamos a utilizar un pipeline llamado nf-core/demo que es mantenido por el proyecto nf-core como parte de su inventario de pipelines para demostración y capacitación.
Asegúrese de que su directorio de trabajo esté configurado en hello-nf-core/ como se indica en la página Primeros pasos.
1. Encontrar y obtener el pipeline nf-core/demo¶
Comencemos localizando el pipeline nf-core/demo en el sitio web del proyecto en nf-co.re, que centraliza toda la información como: documentación general y artículos de ayuda, documentación para cada uno de los pipelines, publicaciones de blog, anuncios de eventos, etcétera.
1.1. Encontrar el pipeline en el sitio web¶
En su navegador web, vaya a https://nf-co.re/pipelines/ y escriba demo en la barra de búsqueda.
Haga clic en el nombre del pipeline, demo, para acceder a la página de documentación del pipeline.
Cada pipeline publicado tiene una página dedicada que incluye las siguientes secciones de documentación:
- Introduction: Una introducción y descripción general del pipeline
- Usage: Descripciones de cómo ejecutar el pipeline
- Parameters: Parámetros del pipeline agrupados con descripciones
- Output: Descripciones y ejemplos de los archivos de salida esperados
- Results: Archivos de salida de ejemplo generados a partir del conjunto de datos de prueba completo
- Releases & Statistics: Historial de versiones del pipeline y estadísticas
Siempre que esté considerando adoptar un nuevo pipeline, debe leer cuidadosamente la documentación del pipeline primero para entender qué hace y cómo debe configurarse antes de intentar ejecutarlo.
Eche un vistazo ahora y vea si puede averiguar:
- Qué herramientas ejecutará el pipeline (Consulte la pestaña:
Introduction) - Qué entradas y parámetros acepta o requiere el pipeline (Consulte la pestaña:
Parameters) - Cuáles son las salidas producidas por el pipeline (Consulte la pestaña:
Output)
1.1.1. Descripción general del pipeline¶
La pestaña Introduction proporciona una descripción general del pipeline, incluyendo una representación visual (llamada mapa de metro) y una lista de herramientas que se ejecutan como parte del pipeline.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
- Generate a lighthearted text message from a cow (COWPY)
1.1.2. Ejemplo de línea de comando¶
La documentación también proporciona un archivo de entrada de ejemplo (discutido más adelante) y un ejemplo de línea de comando.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Notará que el comando de ejemplo NO especifica un archivo de workflow, solo la referencia al repositorio del pipeline, nf-core/demo.
Cuando se invoca de esta manera, Nextflow asumirá que el código está organizado de cierta manera. Obtengamos el código para que podamos examinar esta estructura.
1.2. Obtener el código del pipeline¶
Una vez que hemos determinado que el pipeline parece ser adecuado para nuestros propósitos, probémoslo. Afortunadamente, Nextflow facilita la obtención de pipelines desde repositorios correctamente formateados sin tener que descargar nada manualmente.
1.2.1. Usar nextflow pull¶
Volvamos a la terminal y ejecutemos lo siguiente:
Salida del comando
Nextflow hace un pull del código del pipeline, lo que significa que descarga el repositorio completo en su unidad local.
Para ser claros, puede hacer esto con cualquier pipeline de Nextflow que esté configurado apropiadamente en GitHub, no solo con pipelines de nf-core. Sin embargo, nf-core es la colección de código abierto más grande de pipelines de Nextflow.
1.2.2. Usar nextflow list¶
Puede hacer que Nextflow le proporcione una lista de qué pipelines ha obtenido de esta manera:
Puede intentar obtener algunos otros pipelines para ver cómo aparecen listados cuando tiene más de uno.
1.2.3. Encontrar dónde se descargó el pipeline¶
Notará que los archivos no están en su directorio de trabajo actual.
Por defecto, Nextflow guarda los pipelines obtenidos en $NXF_HOME/assets.
Para encontrar dónde vive un pipeline específico, consulte a Nextflow directamente:
Salida del comando
project name: nf-core/demo
repository : https://github.com/nf-core/demo
local path : /workspaces/.nextflow/assets/.repos/nf-core/demo
main script : main.nf
description : An nf-core demo pipeline
revisions :
TEMPLATE
bumper
dev
fix-nxfversion
manually-merge-3_0_2
> master (default)
nf-core-template-merge-2.13.2.dev0
nf-core-template-merge-2.14.0
nf-core-template-merge-2.14.1
nf-core-template-merge-3.0.0
nf-core-template-merge-3.0.1
nf-core-template-merge-3.0.2
nf-core-template-merge-3.1.0
nf-core-template-merge-3.1.2
nf-core-template-merge-3.2.0
nf-core-template-merge-3.2.1
nf-core-template-merge-3.3.1
nf-core-template-merge-3.3.2
nf-core-template-merge-4.0.0
1.0.0 [t]
1.0.1 [t]
1.0.2 [t]
1.1.0 [t]
> 1.2.0 [t]
Info
La ruta completa puede diferir en su sistema si no está utilizando nuestro entorno de capacitación.
Nextflow mantiene el código fuente descargado intencionalmente 'fuera del camino' bajo el principio de que estos pipelines deben usarse más como bibliotecas que como código con el que interactuaría directamente.
Internamente, Nextflow almacena cada pipeline obtenido como un repositorio git bajo $NXF_HOME/assets/.repos/, y extrae el código de cada revisión en un subdirectorio clones/<commit>/.
Como .repos es un directorio oculto, un simple tree -L 2 $NXF_HOME/assets/ aparecerá vacío.
1.2.4. Crear un enlace simbólico para acceder fácilmente al código fuente¶
No vamos a revisar el código en detalle, pero echemos un vistazo rápido para tener una idea de cómo se ve la organización general.
Para facilitar la exploración del código fuente del pipeline, cree un enlace simbólico que apunte a la copia extraída del pipeline:
mkdir -p pipelines/nf-core
ln -s "$(echo $NXF_HOME/assets/.repos/nf-core/demo/clones/*/)" pipelines/nf-core/demo
Esto crea un acceso directo para que pueda explorar el código con tree -L 2 pipelines/nf-core/demo o abrir archivos directamente.
1.2.5. Descripción general de la organización del código¶
Puede usar tree o usar el explorador de archivos para encontrar y abrir el directorio nf-core/demo.
Contenido del directorio
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
7 directories, 12 files
Como puede ver, hay mucho sucediendo allí, aunque la mayor parte no necesita preocuparle.
Brevemente, notemos que en el nivel superior puede encontrar un archivo README con información resumida, así como archivos accesorios que resumen información del proyecto como licencia, directrices de contribución, citas y código de conducta.
La documentación detallada del pipeline se encuentra en el directorio docs.
Todo este contenido se utiliza para generar las páginas web en el sitio web de nf-core programáticamente, por lo que siempre están actualizadas con el código.
Para el resto, podemos distinguir tres grupos funcionales de archivos de código:
- Componentes del código del pipeline (
main.nf,workflows,subworkflows,modules) - Configuración del pipeline
- Parámetros del pipeline / entradas y validación
No revisaremos los componentes del código del pipeline en esta parte del curso, pero sí tocaremos elementos de configuración y validación que probablemente sean relevantes para usted como usuario final de pipelines de nf-core.
Consejo
También puede explorar el código fuente de cualquier pipeline de nf-core en GitHub, por ejemplo github.com/nf-core/demo. Todos los pipelines de nf-core siguen el mismo diseño de directorios, por lo que una vez que conozca la estructura, puede encontrar archivos de configuración, módulos y workflows de cualquier pipeline de la misma manera.
¡Pero por ahora, pasemos a ejecutar el pipeline!
Conclusión¶
Ahora sabe cómo encontrar un pipeline a través del sitio web de nf-core y obtener una copia local del código fuente.
¿Qué sigue?¶
Aprenda cómo probar un pipeline de nf-core con mínimo esfuerzo.
2. Probar el pipeline con su perfil de prueba¶
Convenientemente, cada pipeline de nf-core viene con un perfil de prueba. Este es un conjunto mínimo de configuraciones para que el pipeline se ejecute usando un conjunto de datos de prueba pequeño alojado en el repositorio nf-core/test-datasets. Es una excelente manera de probar rápidamente un pipeline a pequeña escala.
Consejo
El sistema de perfiles de configuración de Nextflow le permite cambiar fácilmente entre diferentes motores de contenedores o entornos de ejecución. Para más detalles, consulte Hello Nextflow Parte 6: Configuración.
2.1. Examinar el perfil de prueba¶
Es una buena práctica verificar qué especifica el perfil de prueba de un pipeline antes de ejecutarlo.
El perfil test para nf-core/demo se encuentra en el archivo de configuración conf/test.config.
Puede encontrarlo localmente dentro del código fuente del pipeline que descargó nextflow pull, a través del enlace simbólico pipelines creado en la sección 1.2.4:
A continuación se muestra el contenido de ese archivo:
Notará de inmediato que el bloque de comentarios en la parte superior incluye un ejemplo de uso que muestra cómo ejecutar el pipeline con este perfil de prueba.
| conf/test.config | |
|---|---|
Las únicas cosas que necesitamos proporcionar son lo que se muestra entre corchetes angulares en el comando de ejemplo: <docker/singularity> y <OUTDIR>.
Como recordatorio, <docker/singularity> se refiere a la elección del sistema de contenedores. Todos los pipelines de nf-core están diseñados para ser utilizables con contenedores (Docker, Singularity, etc.) para garantizar la reproducibilidad y eliminar problemas de instalación de software.
Entonces necesitaremos especificar si queremos usar Docker o Singularity para probar el pipeline.
La parte --outdir <OUTDIR> se refiere al directorio donde Nextflow escribirá las salidas del pipeline.
Necesitamos proporcionar un nombre para él, que simplemente podemos inventar.
Si aún no existe, Nextflow lo creará por nosotros en tiempo de ejecución.
Pasando a la sección después del bloque de comentarios, el perfil de prueba nos muestra qué ha sido preconfigurado para las pruebas: más notablemente, el parámetro input ya está configurado para apuntar a un conjunto de datos de prueba, por lo que no necesitamos proporcionar nuestros propios datos.
Si sigue el enlace a la entrada preconfigurada, verá que es un archivo CSV que contiene identificadores de muestra y rutas de archivo para varias muestras experimentales.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
Esto se llama una hoja de muestras, y es la forma más común de entrada a los pipelines de nf-core. No se preocupe si no está familiarizado con los formatos y tipos de datos, no es importante para lo que sigue.
Ahora tenemos todo lo que necesitamos para probar el pipeline.
2.2. Ejecutar el pipeline¶
Como se indicó anteriormente, podemos usar el comando de prueba de ejemplo casi tal como está; solo necesitamos especificar qué sistema de empaquetado de software usar y cómo nombrar el directorio de salida.
Aquí usaremos Docker para el sistema de contenedores y demo-results, respectivamente.
Con eso, podemos ejecutar el comando de prueba:
Salida del comando
N E X T F L O W ~ version 26.04.4
Downloading plugin nf-schema@2.7.2
Launching `https://github.com/nf-core/demo` [cranky_curry] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_21-31-35
Core Nextflow options
revision : master
runName : cranky_curry
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Si su salida coincide con esa, ¡felicitaciones! Acaba de ejecutar su primer pipeline de nf-core.
Notará que hay mucha más salida en la consola que cuando ejecuta un pipeline básico de Nextflow. Hay un encabezado que incluye un resumen de la versión del pipeline, entradas y salidas, y algunos elementos de configuración.
Info
Su salida mostrará diferentes marcas de tiempo, nombres de ejecución y rutas de archivo, pero la estructura general y la ejecución del proceso deben ser similares.
Observe la línea cerca de la parte superior de la salida:
Esto le indica qué revisión del pipeline se utilizó.
Como no especificamos una versión, Nextflow utilizó el último commit en master.
Para ejecuciones reproducibles, debe fijar una versión específica usando el indicador -r:
Esto garantiza que se use el mismo código del pipeline cada vez, independientemente de nuevos commits o versiones.
Para esta capacitación omitimos -r por simplicidad, pero en producción siempre debe especificarlo.
Pasando a la salida de ejecución, echemos un vistazo a las líneas que nos dicen qué procesos se ejecutaron:
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Esto nos dice que se ejecutaron cuatro procesos, correspondientes a las cuatro herramientas mostradas en la página de documentación del pipeline en el sitio web de nf-core: FASTQC, SEQTK_TRIM, MULTIQC y COWPY.
Los nombres completos de los procesos como se muestran aquí, como NFCORE_DEMO:DEMO:MULTIQC, son más largos que lo que puede haber visto en el material introductorio de Hello Nextflow.
Estos incluyen los nombres de sus workflows padre y reflejan la modularidad del código del pipeline.
Entraremos en más detalle sobre eso en la Parte 2 de este curso.
2.3. Examinar las salidas del pipeline¶
Finalmente, echemos un vistazo al directorio demo-results producido por el pipeline.
Contenido del directorio
demo-results
├── cowpy
│ └── cowpy.txt
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2026-07-03_21-31-35.html
├── execution_timeline_2026-07-03_21-31-35.html
├── execution_trace_2026-07-03_21-31-35.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2026-07-03_21-31-43.json
└── pipeline_dag_2026-07-03_21-31-35.html
12 directories, 8 files
Eso puede parecer mucho.
Para obtener más información sobre las salidas del pipeline nf-core/demo, consulte su página de documentación.
En esta etapa, lo importante a observar es que los resultados están organizados por módulo, y además hay un directorio llamado pipeline_info que contiene varios informes con marcas de tiempo sobre la ejecución del pipeline.
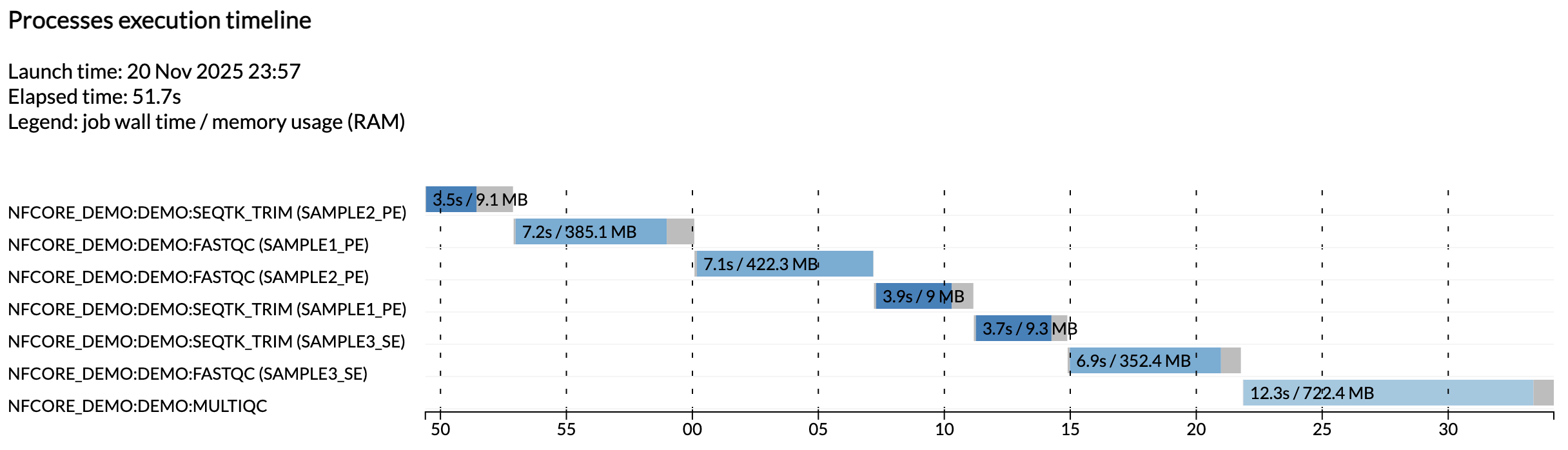
Por ejemplo, el archivo execution_timeline_* le muestra qué procesos se ejecutaron, en qué orden y cuánto tiempo tardaron en ejecutarse:
Info
Aquí las tareas no se ejecutaron en paralelo porque estamos ejecutando en una máquina minimalista en Github Codespaces. Para ver que se ejecuten en paralelo, intente aumentar la asignación de CPU de su codespace y los límites de recursos en la configuración de prueba.
Estos informes se generan automáticamente para todos los pipelines de nf-core.
Conclusión¶
Sabe cómo ejecutar un pipeline de nf-core usando su perfil de prueba integrado y dónde encontrar sus salidas.
¿Qué sigue?¶
Aprenda cómo configurar el pipeline para personalizar su ejecución.
3. Configurar la ejecución del pipeline¶
Como se explica en Hello Config, queremos poder cambiar con qué datos se ejecutará nuestro pipeline y cómo se ejecutará sin modificar el código del pipeline en sí. Para ello, Nextflow admite múltiples formas de controlar la configuración del pipeline, lo que puede resultar un poco abrumador.
El proyecto nf-core especifica convenciones para organizar los elementos de configuración, distinguiendo dos tipos de configuración en el nivel superior: parámetros del pipeline y configuración en sentido estricto.
- Parámetros del pipeline (establecidos a través del sistema
params) típicamente incluyen cosas como archivos de entrada, indicadores de comportamiento de herramientas y parámetros de análisis. - Configuración en sentido estricto se refiere a la logística de cómo se ejecuta el pipeline, es decir, el executor, las asignaciones de recursos de cómputo, etcétera.
Comencemos abordando los parámetros del pipeline, luego veremos la configuración en sentido estricto.
3.1. Parámetros del pipeline¶
Para todos los pipelines de nf-core, puede obtener una lista completa de parámetros del pipeline directamente desde la línea de comando usando el indicador --help, que es en sí mismo un parámetro del pipeline.
3.1.1. Obtener la lista de parámetros con --help¶
Ejecute el comando de ayuda para el pipeline de demostración:
Salida del comando
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [adoring_meucci] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 19 param(s), use the `--showHidden` parameter to show them !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Como puede ver, la salida agrupa los parámetros en categorías (opciones de entrada/salida, opciones de genoma de referencia, etc.) con tipos y descripciones para cada uno.
Esta categorización está determinada por un archivo de esquema, que se trata más adelante.
En pipelines de Nextflow simples, --help solo funciona si el desarrollador lo implementó manualmente.
Consejo
Use --help --show_hidden para ver parámetros adicionales que están ocultos por defecto, como --publish_dir_mode o --monochrome_logs.
3.1.2. Establecer valores de parámetros¶
Como se explica en Hello Config, puede establecer valores de parámetros en la línea de comando con --nombre_param o recopilar un conjunto de parámetros en un archivo YAML y pasarlo con -params-file.
Ambos enfoques funcionan de la misma manera con los pipelines de nf-core.
Por ejemplo, para omitir el paso de recorte, queremos establecer el parámetro boolean skip_trim en true.
En su directorio de trabajo se proporciona un archivo de parámetros llamado my_params.yml con ese valor ya configurado:
Páselo con -params-file:
nextflow run nf-core/demo -profile docker,test --outdir demo-results-notrim -params-file my_params.yml
Salida del comando
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [focused_heisenberg] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results-notrim
Process skipping options
skip_trim : true
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_22-08-47
Core Nextflow options
revision : master
runName : focused_heisenberg
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (5)
[7a/f3599e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b0/2f0bdc] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[c3/3c2278] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
El proceso SEQTK_TRIM ya no aparece en la salida.
Advertencia: Limitaciones importantes sobre las entradas de parámetros
Establecer parámetros boolean en la línea de comando
A partir de la versión 26.04 de Nextflow, todos los valores proporcionados en la línea de comando se tipifican como strings.
Para un parámetro boolean como skip_trim, pasarlo como un indicador simple (--skip_trim) o como --skip_trim true se evalúa como el string "true", lo que falla en la validación del esquema:
Para establecer un parámetro boolean en un valor genuino true/false, use un -params-file como se muestra arriba, o configúrelo en un archivo de configuración.
Los parámetros de tipo string, integer y file-path no se ven afectados y aún pueden establecerse directamente en la línea de comando.
Este curso usa este patrón en todo momento para los parámetros boolean.
Usar archivos de configuración personalizados
Aunque técnicamente es posible establecer parámetros del pipeline en un archivo de configuración personalizado pasado con -c, esto puede no anular los valores predeterminados ya establecidos en el propio nextflow.config del pipeline, dependiendo de las reglas de precedencia de configuración de Nextflow.
Usar --nombre_param en la línea de comando o -params-file es más confiable, ya que estos siempre tienen precedencia.
Como regla general: si aparece en la salida de --help, establézcalo a través de la línea de comando o un archivo de parámetros en lugar de un archivo de configuración.
3.1.3. Validación de parámetros¶
Dato curioso: el comando --help funciona para todos los pipelines de nf-core porque el proyecto nf-core requiere que los desarrolladores definan formalmente todos los parámetros del pipeline en un archivo de esquema JSON (nextflow_schema.json).
Este esquema registra el tipo, descripción, valor predeterminado y agrupación de cada parámetro.
Además de potenciar la salida de --help, el archivo de esquema también permite la validación automatizada en el momento del lanzamiento.
Esto significa que Nextflow puede verificar que cada parámetro que pase exista y haya recibido un valor apropiado (del tipo apropiado, dentro del rango de valores permitidos, etc.).
Cubrimos esto con más detalle en Parte 5: Validación de entrada, pero ya puede verlo en acción proporcionando al pipeline de demostración alguna entrada de parámetros no válida.
3.1.3.1. Parámetros no reconocidos¶
Intente pasar un parámetro que no existe:
La salida de la consola incluye una advertencia:
El pipeline sigue ejecutándose, pero la advertencia le alerta de inmediato que --foobar no es un parámetro reconocido.
Esto está diseñado para llamar su atención sobre errores tipográficos que no interrumpen la ejecución, como usar --outDir en lugar de --outdir, lo que puede ayudarle a evitar perder tiempo y recursos de cómputo.
3.1.3.2. Valores de parámetros no válidos¶
La validación también verifica los valores de los parámetros.
El parámetro --skip_trim es un indicador boolean, por lo que pasar un valor de tipo string hace que el pipeline falle inmediatamente:
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
El pipeline se detiene antes de que se ejecute cualquier proceso, evitando una ejecución fallida o incorrecta.
Como se indica en la sección 3.1.2, los parámetros boolean deben establecerse en un valor genuino true/false en un archivo de parámetros en lugar de pasarse en la línea de comando, ya que los valores de la línea de comando se tipifican como strings.
3.1.4. Validación de entrada¶
La misma lógica de validación también puede usarse para verificar la validez de los archivos de entrada. Por ejemplo, si un pipeline espera una hoja de muestras como su entrada de datos principal (que es el caso de muchos si no la mayoría de los pipelines de nf-core), el desarrollador puede proporcionar un esquema de entrada (distinto del esquema de parámetros) que describa cómo debe estar estructurado el archivo de entrada.
Luego, en tiempo de ejecución, Nextflow puede verificar que el archivo de entrada proporcionado sea válido.
También cubrimos esto con más detalle en Parte 5: Validación de entrada, pero ya puede verlo en acción proporcionando al pipeline de demostración una hoja de muestras de entrada no válida.
El pipeline nf-core/demo espera un archivo CSV con columnas sample, fastq_1 y fastq_2.
Esto está definido en un archivo de esquema (assets/schema_input.json) que especifica la estructura esperada, los tipos de columnas y las restricciones.
Archivo de esquema para las entradas
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
El esquema especifica que sample y fastq_1 son obligatorios, mientras que fastq_2 es opcional (admitiendo datos de extremo pareado y de extremo único).
Las rutas de archivo se validan para verificar su existencia y patrón de extensión.
Para demostrarlo, en su directorio de trabajo se proporciona una hoja de muestras malformada llamada malformed_samplesheet.csv:
A esta hoja de muestras le falta la columna requerida fastq_1 y tiene una ruta de archivo inexistente en fastq_2.
Ejecute el pipeline de demostración usando malformed_samplesheet.csv como entrada:
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Como puede ver, el pipeline falla inmediatamente e informa todos los errores de validación a la vez. nf-schema no se detiene en el primer error — recopila todos los problemas y los lista juntos, para que pueda corregirlos todos de una vez en lugar de descubrir los problemas uno por uno.
Cada error identifica la entrada y el campo exactos que causaron el problema, para que pueda corregir su hoja de muestras y luego volver a lanzar el pipeline con la confianza de que no va a fallar en algún punto posterior cuando Nextflow intente acceder a la ruta del archivo.
Para los desarrolladores, todo esto se cubre con más detalle en la Parte 5 de este curso.
3.2. Configuración¶
La configuración en sentido estricto controla cómo se ejecuta el pipeline: asignación de recursos, argumentos específicos de herramientas, dónde se ejecutan las tareas y qué sistema de empaquetado de software se usa.
Los pipelines de nf-core incluyen configuración predeterminada en nextflow.config y el directorio conf/.
Antes de anular cualquier cosa, es útil saber dónde se encuentran los valores predeterminados.
Ya vio en la sección 2.1 que el código fuente del pipeline se encuentra bajo $NXF_HOME/assets.
Usando el enlace simbólico pipelines de la sección 1.2.4, liste los archivos de configuración para ver qué está disponible:
base.config
containers_conda_lock_files_amd64.config
containers_conda_lock_files_arm64.config
containers_docker_amd64.config
containers_docker_arm64.config
containers_singularity_https_amd64.config
containers_singularity_https_arm64.config
containers_singularity_oras_amd64.config
containers_singularity_oras_arm64.config
igenomes.config
igenomes_ignored.config
modules.config
test.config
test_full.config
Los archivos de configuración más importantes son:
conf/base.config: Define etiquetas de recursos (process_low,process_medium,process_high) que asignan CPUs, memoria y tiempo a los procesos. Cuando vea que un proceso usa más recursos de los esperados, aquí es donde se originan esos valores predeterminados.conf/modules.config: Establece argumentos de herramientas por proceso (ext.args) y configuraciones de publicación de salidas (publishDir). Abra este archivo para ver qué argumentos recibe cada herramienta por defecto.conf/test.config: El perfil de prueba que usó en la sección 2.1, que limita los recursos medianteresourceLimitsy establece una hoja de muestras de prueba. Se activa con-profile test. También hay unconf/test_full.configpara ejecutar con un conjunto de datos de prueba de tamaño completo, útil para benchmarking.
El nextflow.config central carga todos los anteriores y establece los valores predeterminados apropiados para todo.
Si desea modificar alguna de las configuraciones especificadas en estos archivos, no modifique ninguno de ellos directamente.
En su lugar, cree su propio archivo de configuración y páselo con -c.
Los valores que especifique anularán los valores predeterminados establecidos en esos otros archivos.
Practiquemos esto.
3.2.1. Personalizar recursos de procesos y argumentos de herramientas¶
Los módulos de nf-core admiten dos tipos comunes de anulación de configuración: asignación de recursos (CPUs, memoria, tiempo) y argumentos de herramientas a través de ext.args.
Muchas herramientas de línea de comando tienen argumentos que no se usan con suficiente frecuencia como para exponerse como parámetros del pipeline.
La convención ext.args le permite pasar estos argumentos a la herramienta subyacente a través de un archivo de configuración.
El archivo custom.config proporcionado en su directorio de trabajo demuestra ambas anulaciones:
| custom.config | |
|---|---|
El primer bloque anula la asignación de recursos de FASTQC.
Por defecto, FASTQC usa la etiqueta process_medium de base.config, que asigna 6 CPUs y 36 GB de memoria; aquí lo limitamos a 2 CPUs y 4 GB.
El segundo bloque pasa un argumento adicional a SEQTK_TRIM a través de ext.args.
El indicador -b 5 le indica a seqtk trimfq que recorte 5 bases del inicio de cada lectura además del recorte por calidad.
Ejecute el pipeline con esta configuración:
Salida del comando
El indicador -c agrega su configuración sobre la configuración integrada del pipeline.
Para verificar que la anulación de ext.args tuvo efecto, encuentre el hash del directorio de trabajo de SEQTK_TRIM en la salida de la ejecución (por ejemplo, work/17/428668...) y verifique el archivo .command.sh dentro de él:
Salida del comando
Debería ver -b 5 en el comando seqtk trimfq.
Una cosa importante que debe saber sobre ext.args: si un módulo ya tiene un valor predeterminado establecido, su valor lo reemplazará completamente en lugar de agregarse a él.
Por ejemplo, FASTQC tiene ext.args = '--quiet' establecido por defecto en conf/modules.config:
| conf/modules.config | |
|---|---|
Si establece ext.args = '--kmers 8' para FASTQC, el indicador --quiet ya no se aplicará.
Para mantener ambos, establezca ext.args = '--quiet --kmers 8'.
Siempre debe verificar la configuración predeterminada de un módulo antes de anular ext.args.
Conclusión¶
Sabe cómo obtener ayuda de un pipeline de nf-core, establecer parámetros y entender cómo se validan, y personalizar la configuración a través de archivos de configuración.
¿Qué sigue?¶
¡Si solo quiere ejecutar pipelines de nf-core, ya terminó!
Si quiere aprender a desarrollar sus propios pipelines según los estándares de nf-core, tome un descanso y pase a la Parte 2 cuando esté listo. Aprenderá a crear su propio pipeline compatible con nf-core usando las herramientas basadas en la plantilla de nf-core.