Partie 1 : Exécuter un pipeline de démonstration¶
Traduction assistée par IA - en savoir plus et suggérer des améliorations
Dans cette première partie de la formation Hello nf-core, nous vous montrons comment trouver et essayer un pipeline nf-core, configurer et personnaliser son exécution selon vos besoins, et comprendre comment la validation des entrées protège contre les erreurs courantes.
Nous allons utiliser un pipeline appelé nf-core/demo qui est maintenu par le projet nf-core dans le cadre de son inventaire de pipelines à des fins de démonstration et de formation.
Assurez-vous que votre répertoire de travail est défini sur hello-nf-core/ comme indiqué sur la page Premiers pas.
1. Trouver et récupérer le pipeline nf-core/demo¶
Commençons par localiser le pipeline nf-core/demo sur le site web du projet à l'adresse nf-co.re, qui centralise toutes les informations telles que : la documentation générale et les articles d'aide, la documentation pour chacun des pipelines, les articles de blog, les annonces d'événements, etc.
1.1. Trouver le pipeline sur le site web¶
Dans votre navigateur web, allez sur https://nf-co.re/pipelines/ et tapez demo dans la barre de recherche.
Cliquez sur le nom du pipeline, demo, pour accéder à la page de documentation du pipeline.
Chaque pipeline publié dispose d'une page dédiée qui comprend les sections de documentation suivantes :
- Introduction : Une introduction et un aperçu du pipeline
- Usage : Descriptions de la manière d'exécuter le pipeline
- Parameters : Paramètres du pipeline regroupés avec descriptions
- Output : Descriptions et exemples des fichiers de sortie attendus
- Results : Exemples de fichiers de sortie générés à partir du jeu de données de test complet
- Releases & Statistics : Historique des versions du pipeline et statistiques
Chaque fois que vous envisagez d'adopter un nouveau pipeline, vous devez d'abord lire attentivement la documentation du pipeline pour comprendre ce qu'il fait et comment il doit être configuré avant de tenter de l'exécuter.
Jetez-y un coup d'œil maintenant et voyez si vous pouvez découvrir :
- Quels outils le pipeline exécutera (Vérifiez l'onglet :
Introduction) - Quelles entrées et paramètres le pipeline accepte ou requiert (Vérifiez l'onglet :
Parameters) - Quelles sont les sorties produites par le pipeline (Vérifiez l'onglet :
Output)
1.1.1. Aperçu du pipeline¶
L'onglet Introduction fournit un aperçu du pipeline, incluant une représentation visuelle (appelée carte de métro) et une liste des outils qui sont exécutés dans le cadre du pipeline.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
- Generate a lighthearted text message from a cow (COWPY)
1.1.2. Exemple de ligne de commande¶
La documentation fournit également un exemple de fichier d'entrée (discuté plus en détail ci-dessous) et un exemple de ligne de commande.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Vous remarquerez que l'exemple de commande ne spécifie PAS de fichier de workflow, juste la référence au dépôt du pipeline, nf-core/demo.
Lorsqu'il est invoqué de cette manière, Nextflow supposera que le code est organisé d'une certaine manière. Récupérons le code afin de pouvoir examiner cette structure.
1.2. Récupérer le code du pipeline¶
Une fois que nous avons déterminé que le pipeline semble convenir à nos besoins, essayons-le. Heureusement, Nextflow facilite la récupération des pipelines à partir de dépôts correctement formatés sans avoir à télécharger quoi que ce soit manuellement.
1.2.1. Utiliser nextflow pull¶
Retournons au terminal et exécutons ce qui suit :
Sortie de la commande
Nextflow effectue un pull du code du pipeline, c'est-à-dire qu'il télécharge le dépôt complet sur votre disque local.
Pour être clair, vous pouvez faire cela avec n'importe quel pipeline Nextflow qui est correctement configuré dans GitHub, pas seulement les pipelines nf-core. Cependant, nf-core est la plus grande collection open-source de pipelines Nextflow.
1.2.2. Utiliser nextflow list¶
Vous pouvez demander à Nextflow de vous fournir une liste des pipelines que vous avez récupérés de cette manière :
Vous pouvez essayer de récupérer quelques autres pipelines pour voir comment ils apparaissent dans la liste lorsque vous en avez plusieurs.
1.2.3. Trouver l'emplacement du pipeline téléchargé¶
Vous remarquerez que les fichiers ne se trouvent pas dans votre répertoire de travail actuel.
Par défaut, Nextflow enregistre les pipelines récupérés dans $NXF_HOME/assets.
Pour trouver l'emplacement d'un pipeline spécifique, interrogez Nextflow directement :
Sortie de la commande
project name: nf-core/demo
repository : https://github.com/nf-core/demo
local path : /workspaces/.nextflow/assets/.repos/nf-core/demo
main script : main.nf
description : An nf-core demo pipeline
revisions :
TEMPLATE
bumper
dev
fix-nxfversion
manually-merge-3_0_2
> master (default)
nf-core-template-merge-2.13.2.dev0
nf-core-template-merge-2.14.0
nf-core-template-merge-2.14.1
nf-core-template-merge-3.0.0
nf-core-template-merge-3.0.1
nf-core-template-merge-3.0.2
nf-core-template-merge-3.1.0
nf-core-template-merge-3.1.2
nf-core-template-merge-3.2.0
nf-core-template-merge-3.2.1
nf-core-template-merge-3.3.1
nf-core-template-merge-3.3.2
nf-core-template-merge-4.0.0
1.0.0 [t]
1.0.1 [t]
1.0.2 [t]
1.1.0 [t]
> 1.2.0 [t]
Info
Le chemin complet peut différer sur votre système si vous n'utilisez pas notre environnement de formation.
Nextflow garde intentionnellement le code source téléchargé 'à l'écart' sur le principe que ces pipelines doivent être utilisés davantage comme des bibliothèques que comme du code avec lequel vous interagiriez directement.
En coulisses, Nextflow stocke chaque pipeline récupéré comme un dépôt git dans $NXF_HOME/assets/.repos/, et extrait le code pour chaque révision dans un sous-répertoire clones/<commit>/.
Comme .repos est un répertoire caché, un simple tree -L 2 $NXF_HOME/assets/ semblera vide.
1.2.4. Créer un lien symbolique pour accéder facilement au code source¶
Nous n'allons pas examiner le code en détail, mais jetons-y un coup d'œil rapide pour avoir une idée de l'organisation générale.
Pour faciliter la navigation dans le code source du pipeline, créez un lien symbolique pointant vers la copie extraite du pipeline :
mkdir -p pipelines/nf-core
ln -s "$(echo $NXF_HOME/assets/.repos/nf-core/demo/clones/*/)" pipelines/nf-core/demo
Cela crée un raccourci qui vous permet d'explorer le code avec tree -L 2 pipelines/nf-core/demo ou d'ouvrir des fichiers directement.
1.2.5. Aperçu de l'organisation du code¶
Vous pouvez soit utiliser tree soit utiliser l'explorateur de fichiers pour trouver et ouvrir le répertoire nf-core/demo.
Contenu du répertoire
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
7 directories, 12 files
Comme vous pouvez le voir, il se passe beaucoup de choses là-dedans, dont la plupart ne vous concernent pas.
Notons brièvement qu'au niveau supérieur, vous pouvez trouver un fichier README avec des informations récapitulatives, ainsi que des fichiers accessoires qui résument les informations du projet telles que les licences, les directives de contribution, les citations et le code de conduite.
La documentation détaillée du pipeline se trouve dans le répertoire docs.
Tout ce contenu est utilisé pour générer les pages web sur le site web nf-core de manière programmatique, elles sont donc toujours à jour avec le code.
Pour le reste, nous pouvons distinguer trois groupes fonctionnels de fichiers de code :
- Composants du code du pipeline (
main.nf,workflows,subworkflows,modules) - Configuration du pipeline
- Paramètres du pipeline / entrées et validation
Nous ne passerons pas en revue les composants du code du pipeline dans cette partie du cours, mais nous aborderons les éléments de configuration et de validation qui vous seront probablement utiles en tant qu'utilisateur·trice final·e de pipelines nf-core.
Astuce
Vous pouvez également parcourir le code source de n'importe quel pipeline nf-core sur GitHub, par exemple github.com/nf-core/demo. Chaque pipeline nf-core suit la même organisation de répertoires, donc une fois que vous connaissez la structure, vous pouvez trouver les fichiers de configuration, les modules et les workflows de n'importe quel pipeline de la même manière.
Mais pour l'instant, passons à l'exécution du pipeline !
À retenir¶
Vous savez maintenant comment trouver un pipeline via le site web nf-core et récupérer une copie locale du code source.
Et ensuite ?¶
Apprenez comment essayer un pipeline nf-core avec un minimum d'effort.
2. Essayer le pipeline avec son profil de test¶
De manière pratique, chaque pipeline nf-core est fourni avec un profil de test. Il s'agit d'un ensemble minimal de paramètres de configuration permettant au pipeline de s'exécuter en utilisant un petit jeu de données de test hébergé dans le dépôt nf-core/test-datasets. C'est un excellent moyen d'essayer rapidement un pipeline à petite échelle.
Astuce
Le système de profils de configuration de Nextflow vous permet de basculer facilement entre différents moteurs de conteneurs ou environnements d'exécution. Pour plus de détails, consultez Hello Nextflow Partie 6 : Configuration.
2.1. Examiner le profil de test¶
C'est une bonne pratique de vérifier ce que spécifie le profil de test d'un pipeline avant de l'exécuter.
Le profil test pour nf-core/demo se trouve dans le fichier de configuration conf/test.config.
Vous pouvez le trouver localement dans le code source du pipeline téléchargé par nextflow pull, via le lien symbolique pipelines créé à la section 1.2.4 :
Voici le contenu de ce fichier :
Vous remarquerez tout de suite que le bloc de commentaires en haut inclut un exemple d'utilisation montrant comment exécuter le pipeline avec ce profil de test.
| conf/test.config | |
|---|---|
Les seules choses que nous devons fournir sont ce qui est montré entre crochets dans l'exemple de commande : <docker/singularity> et <OUTDIR>.
Pour rappel, <docker/singularity> fait référence au choix du système de conteneurs. Tous les pipelines nf-core sont conçus pour être utilisables avec des conteneurs (Docker, Singularity, etc.) afin de garantir la reproductibilité et d'éliminer les problèmes d'installation de logiciels.
Nous devrons donc spécifier si nous voulons utiliser Docker ou Singularity pour tester le pipeline.
La partie --outdir <OUTDIR> fait référence au répertoire où Nextflow écrira les sorties du pipeline.
Nous devons lui fournir un nom, que nous pouvons simplement inventer.
S'il n'existe pas déjà, Nextflow le créera pour nous lors de l'exécution.
Passant à la section après le bloc de commentaires, le profil de test nous montre ce qui a été préconfiguré pour les tests : plus particulièrement, le paramètre input est déjà défini pour pointer vers un jeu de données de test, nous n'avons donc pas besoin de fournir nos propres données.
Si vous suivez le lien vers l'entrée préconfigurée, vous verrez qu'il s'agit d'un fichier csv contenant des identifiants d'échantillons et des chemins de fichiers pour plusieurs échantillons expérimentaux.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
C'est ce qu'on appelle une feuille d'échantillons (samplesheet), et c'est la forme d'entrée la plus courante pour les pipelines nf-core. Ne vous inquiétez pas si vous n'êtes pas familier·ère avec les formats et types de données, ce n'est pas important pour la suite.
Nous avons maintenant tout ce dont nous avons besoin pour essayer le pipeline.
2.2. Exécuter le pipeline¶
Comme indiqué ci-dessus, nous pouvons utiliser l'exemple de commande de test presque tel quel ; nous devons simplement spécifier quel système de packaging logiciel utiliser et quel nom donner au répertoire de sortie.
Nous utiliserons Docker pour le système de conteneurs et demo-results, respectivement.
Avec cela, nous pouvons exécuter la commande de test :
Sortie de la commande
N E X T F L O W ~ version 26.04.4
Downloading plugin nf-schema@2.7.2
Launching `https://github.com/nf-core/demo` [cranky_curry] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_21-31-35
Core Nextflow options
revision : master
runName : cranky_curry
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Si votre sortie correspond à celle-ci, félicitations ! Vous venez d'exécuter votre premier pipeline nf-core.
Vous remarquerez qu'il y a beaucoup plus de sortie console que lorsque vous exécutez un pipeline Nextflow basique. Il y a un en-tête qui inclut un résumé de la version du pipeline, des entrées et sorties, et quelques éléments de configuration.
Info
Votre sortie affichera des horodatages, des noms d'exécution et des chemins de fichiers différents, mais la structure globale et l'exécution des processus devraient être similaires.
Remarquez la ligne en haut de la sortie :
Cela vous indique quelle révision du pipeline a été utilisée.
Comme nous n'avons pas spécifié de version, Nextflow a utilisé le dernier commit sur master.
Pour des exécutions reproductibles, vous devriez fixer une version spécifique en utilisant le flag -r :
Cela garantit que le même code de pipeline est utilisé à chaque fois, indépendamment des nouveaux commits ou versions.
Pour cette formation, nous omettons -r par souci de simplicité, mais en production vous devriez toujours le spécifier.
Passons maintenant à la sortie d'exécution, et jetons un coup d'œil aux lignes qui nous indiquent quels processus ont été exécutés :
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Cela nous indique que quatre processus ont été exécutés, correspondant aux quatre outils présentés dans la page de documentation du pipeline sur le site web nf-core : FASTQC, SEQTK_TRIM, MULTIQC et COWPY.
Les noms complets des processus tels qu'affichés ici, comme NFCORE_DEMO:DEMO:MULTIQC, sont plus longs que ce que vous avez pu voir dans le matériel d'introduction Hello Nextflow.
Ils incluent les noms de leurs workflows parents et reflètent la modularité du code du pipeline.
Nous entrerons plus en détail à ce sujet dans la Partie 2 de ce cours.
2.3. Examiner les sorties du pipeline¶
Enfin, jetons un coup d'œil au répertoire demo-results produit par le pipeline.
Contenu du répertoire
demo-results
├── cowpy
│ └── cowpy.txt
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2026-07-03_21-31-35.html
├── execution_timeline_2026-07-03_21-31-35.html
├── execution_trace_2026-07-03_21-31-35.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2026-07-03_21-31-43.json
└── pipeline_dag_2026-07-03_21-31-35.html
12 directories, 8 files
Cela peut sembler beaucoup.
Pour en savoir plus sur les sorties du pipeline nf-core/demo, consultez sa page de documentation.
À ce stade, ce qui est important d'observer est que les résultats sont organisés par module, et il y a en plus un répertoire appelé pipeline_info contenant divers rapports horodatés sur l'exécution du pipeline.
Par exemple, le fichier execution_timeline_* vous montre quels processus ont été exécutés, dans quel ordre et combien de temps ils ont pris pour s'exécuter :
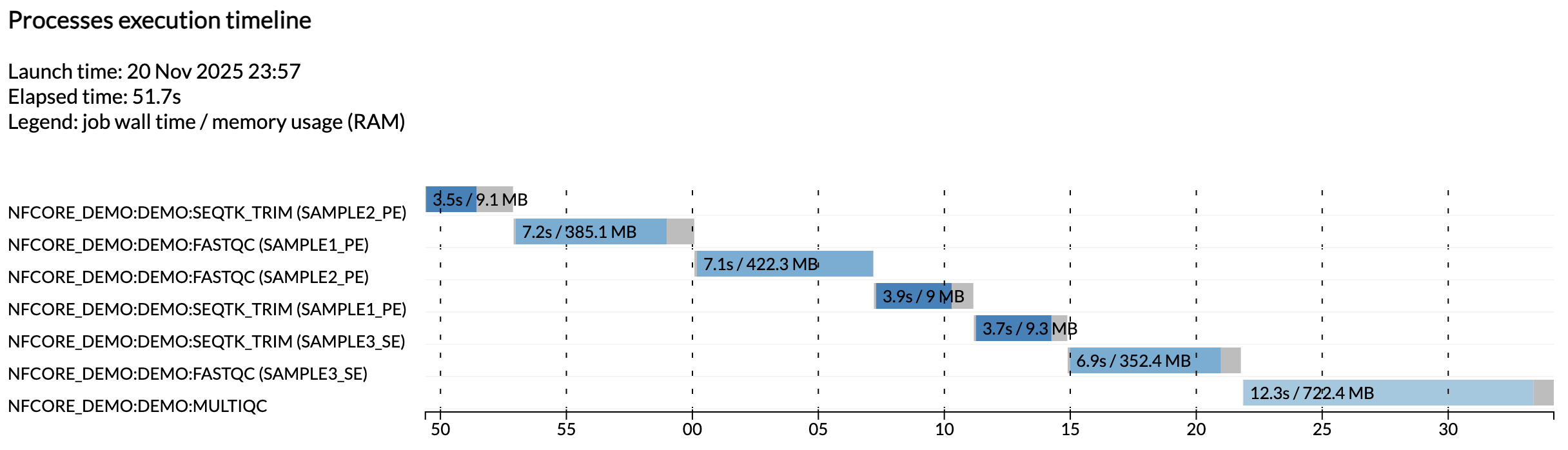
Info
Ici, les tâches n'ont pas été exécutées en parallèle car nous fonctionnons sur une machine minimaliste dans Github Codespaces. Pour voir ces tâches s'exécuter en parallèle, essayez d'augmenter l'allocation CPU de votre codespace et les limites de ressources dans la configuration de test.
Ces rapports sont générés automatiquement pour tous les pipelines nf-core.
À retenir¶
Vous savez comment exécuter un pipeline nf-core en utilisant son profil de test intégré et où trouver ses sorties.
Et ensuite ?¶
Apprenez comment configurer le pipeline pour personnaliser son exécution.
3. Configurer l'exécution du pipeline¶
Comme expliqué dans Hello Config, nous souhaitons pouvoir modifier les données sur lesquelles notre pipeline s'exécutera et la manière dont il s'exécutera sans modifier le code du pipeline lui-même. À cette fin, Nextflow prend en charge plusieurs façons de contrôler la configuration du pipeline, ce qui peut être un peu déroutant.
Le projet nf-core spécifie des conventions pour organiser les éléments de configuration, distinguant deux types de configuration au niveau supérieur : les paramètres du pipeline et la configuration au sens strict.
- Les paramètres du pipeline (définis via le système
params) comprennent généralement des éléments tels que les fichiers d'entrée, les options de comportement des outils et les paramètres d'analyse. - La configuration au sens strict fait référence à la logistique de la façon dont le pipeline est exécuté, c'est-à-dire l'executor, les allocations de ressources de calcul, etc.
Commençons par aborder les paramètres du pipeline, puis nous examinerons la configuration au sens strict.
3.1. Paramètres du pipeline¶
Pour tous les pipelines nf-core, vous pouvez obtenir une liste complète des paramètres du pipeline directement depuis la ligne de commande en utilisant le flag --help, qui est lui-même un paramètre du pipeline.
3.1.1. Obtenir la liste des paramètres avec --help¶
Exécutez la commande d'aide pour le pipeline demo :
Sortie de la commande
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [adoring_meucci] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 19 param(s), use the `--showHidden` parameter to show them !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Comme vous pouvez le voir, la sortie regroupe les paramètres par catégories (options d'entrée/sortie, options de génome de référence, etc.) avec les types et descriptions pour chacun.
Cette catégorisation est déterminée par un fichier de schéma, abordé plus loin ci-dessous.
Dans les pipelines Nextflow simples, --help ne fonctionne que si le développeur l'a implémenté manuellement.
Astuce
Utilisez --help --show_hidden pour voir les paramètres supplémentaires qui sont masqués par défaut, tels que --publish_dir_mode ou --monochrome_logs.
3.1.2. Définir les valeurs des paramètres¶
Comme abordé dans Hello Config, vous pouvez définir les valeurs des paramètres sur la ligne de commande avec --nom_param ou regrouper un ensemble de paramètres dans un fichier YAML et le passer avec -params-file.
Les deux approches fonctionnent de la même manière avec les pipelines nf-core.
Par exemple, pour ignorer l'étape de rognage, nous souhaitons définir le paramètre booléen skip_trim à true.
Un fichier de paramètres appelé my_params.yml est fourni dans votre répertoire de travail avec cette valeur déjà définie :
Passez-le avec -params-file :
nextflow run nf-core/demo -profile docker,test --outdir demo-results-notrim -params-file my_params.yml
Sortie de la commande
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [focused_heisenberg] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results-notrim
Process skipping options
skip_trim : true
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_22-08-47
Core Nextflow options
revision : master
runName : focused_heisenberg
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (5)
[7a/f3599e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b0/2f0bdc] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[c3/3c2278] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Le processus SEQTK_TRIM n'apparaît plus dans la sortie.
Avertissement : limitations importantes concernant les entrées de paramètres
Définir des paramètres booléens sur la ligne de commande
À partir de la version 26.04 de Nextflow, toutes les valeurs fournies sur la ligne de commande sont typées comme des chaînes de caractères.
Pour un paramètre booléen comme skip_trim, le passer comme un flag seul (--skip_trim) ou comme --skip_trim true est évalué comme la chaîne "true", ce qui échoue à la validation du schéma :
Pour définir un paramètre booléen à une valeur true/false authentique, utilisez un -params-file comme indiqué ci-dessus, ou définissez-le dans un fichier de configuration.
Les paramètres de type string, integer et file-path ne sont pas affectés et peuvent toujours être définis directement sur la ligne de commande.
Ce cours utilise ce modèle tout au long pour les paramètres booléens.
Utiliser des fichiers de configuration personnalisés
Bien qu'il soit techniquement possible de définir des paramètres du pipeline dans un fichier de configuration personnalisé passé avec -c, cela peut ne pas remplacer les valeurs par défaut déjà définies dans le propre nextflow.config du pipeline, selon les règles de priorité de configuration de Nextflow.
L'utilisation de --nom_param sur la ligne de commande ou de -params-file est plus fiable, car ces méthodes ont toujours la priorité.
En règle générale : si un paramètre apparaît dans la sortie de --help, définissez-le via la ligne de commande ou un fichier de paramètres plutôt que dans un fichier de configuration.
3.1.3. Validation des paramètres¶
Fait intéressant : la commande --help fonctionne pour tous les pipelines nf-core parce que le projet nf-core exige que les développeur·ses définissent formellement tous les paramètres du pipeline dans un fichier de schéma JSON (nextflow_schema.json).
Ce schéma enregistre le type, la description, la valeur par défaut et le regroupement de chaque paramètre.
En plus d'alimenter la sortie de --help, le fichier de schéma permet également une validation automatisée au moment du lancement.
Cela signifie que Nextflow peut vérifier que chaque paramètre que vous passez existe et a reçu une valeur appropriée (du type approprié, dans la plage de valeurs autorisées, etc.).
Nous abordons cela plus en détail dans la Partie 5 : Validation des entrées, mais vous pouvez déjà le voir en action en fournissant au pipeline demo des entrées de paramètres invalides.
3.1.3.1. Paramètres non reconnus¶
Essayez de passer un paramètre qui n'existe pas :
La sortie console inclut un avertissement :
Le pipeline s'exécute quand même, mais l'avertissement vous alerte immédiatement que --foobar n'est pas un paramètre reconnu.
Cela permet de détecter les fautes de frappe comme --outDir au lieu de --outdir, ce qui peut vous éviter de perdre du temps et des ressources de calcul.
3.1.3.2. Valeurs de paramètres invalides¶
La validation vérifie également les valeurs des paramètres.
Le paramètre --skip_trim est un flag booléen, donc passer une valeur de type string provoque l'échec immédiat du pipeline :
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
Le pipeline s'arrête avant qu'aucun processus ne s'exécute, vous évitant une exécution échouée ou incorrecte.
Comme indiqué à la section 3.1.2, les paramètres booléens doivent être définis à une valeur true/false authentique dans un fichier de paramètres plutôt que passés sur la ligne de commande, car les valeurs en ligne de commande sont typées comme des chaînes de caractères.
3.1.4. Validation des entrées¶
La même logique de validation peut également être utilisée pour vérifier la validité des fichiers d'entrée. Par exemple, si un pipeline attend une feuille d'échantillons comme entrée principale de données (ce qui est le cas de nombreux pipelines nf-core, voire de la plupart), le développeur peut fournir un schéma d'entrée (distinct du schéma des paramètres) décrivant comment le fichier d'entrée doit être structuré.
Ensuite, au moment de l'exécution, Nextflow peut vérifier que le fichier d'entrée fourni est valide.
Nous abordons également cela plus en détail dans la Partie 5 : Validation des entrées, mais vous pouvez déjà le voir en action en fournissant au pipeline demo une feuille d'échantillons invalide.
Le pipeline nf-core/demo attend un fichier CSV avec les colonnes sample, fastq_1 et fastq_2.
Cela est défini dans un fichier de schéma (assets/schema_input.json) qui spécifie la structure attendue, les types de colonnes et les contraintes.
Fichier de schéma pour les entrées
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
Le schéma spécifie que sample et fastq_1 sont obligatoires, tandis que fastq_2 est optionnel (prenant en charge les données paired-end et single-end).
Les chemins de fichiers sont validés pour leur existence et leur extension.
Pour illustrer cela, une feuille d'échantillons malformée appelée malformed_samplesheet.csv est fournie dans votre répertoire de travail :
Cette feuille d'échantillons est dépourvue de la colonne obligatoire fastq_1 et contient un chemin de fichier inexistant dans fastq_2.
Exécutez le pipeline demo en utilisant malformed_samplesheet.csv comme entrée :
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Comme vous pouvez le voir, le pipeline échoue immédiatement et signale toutes les erreurs de validation en même temps. nf-schema ne s'arrête pas à la première erreur — il collecte tous les problèmes et les liste ensemble, afin que vous puissiez tout corriger en une seule fois plutôt que de découvrir les problèmes un par un.
Chaque erreur identifie l'entrée et le champ exacts qui ont causé le problème, afin que vous puissiez corriger votre feuille d'échantillons puis relancer le pipeline avec la certitude qu'il ne va pas échouer à un moment ultérieur lorsque Nextflow tentera d'accéder au chemin du fichier.
Pour les développeur·ses, tout cela est abordé plus en détail dans la Partie 5 de ce cours.
3.2. Configuration¶
La configuration au sens strict contrôle comment le pipeline s'exécute : l'allocation des ressources, les arguments spécifiques aux outils, l'endroit où les tâches s'exécutent et le système de packaging logiciel à utiliser.
Les pipelines nf-core incluent une configuration par défaut dans nextflow.config et le répertoire conf/.
Avant de remplacer quoi que ce soit, il est utile de savoir où se trouvent les valeurs par défaut.
Vous avez déjà vu à la section 2.1 que le code source du pipeline se trouve dans $NXF_HOME/assets.
En utilisant le lien symbolique pipelines de la section 1.2.4, listez les fichiers de configuration pour voir ce qui est disponible :
base.config
containers_conda_lock_files_amd64.config
containers_conda_lock_files_arm64.config
containers_docker_amd64.config
containers_docker_arm64.config
containers_singularity_https_amd64.config
containers_singularity_https_arm64.config
containers_singularity_oras_amd64.config
containers_singularity_oras_arm64.config
igenomes.config
igenomes_ignored.config
modules.config
test.config
test_full.config
Les fichiers de configuration les plus importants sont :
conf/base.config: Définit des labels de ressources (process_low,process_medium,process_high) qui attribuent des CPUs, de la mémoire et du temps aux processus. Lorsque vous constatez qu'un processus utilise plus de ressources que prévu, c'est là que se trouvent ces valeurs par défaut.conf/modules.config: Définit les arguments des outils par processus (ext.args) et les paramètres de publication des sorties (publishDir). Ouvrez ce fichier pour voir quels arguments chaque outil reçoit par défaut.conf/test.config: Le profil de test que vous avez utilisé à la section 2.1, qui limite les ressources viaresourceLimitset définit une feuille d'échantillons de test. Activé avec-profile test. Il existe également unconf/test_full.configpour exécuter le pipeline avec un jeu de données de test de taille complète, utile pour les benchmarks.
Le fichier central nextflow.config charge tous les fichiers ci-dessus et définit les valeurs par défaut appropriées pour tout.
Si vous souhaitez modifier l'un des paramètres spécifiés dans ces fichiers, ne modifiez aucun d'entre eux directement.
Créez plutôt votre propre fichier de configuration et passez-le avec -c.
Les valeurs que vous spécifiez remplaceront les valeurs par défaut définies dans ces autres fichiers.
Essayons cela en pratique.
3.2.1. Personnaliser les ressources des processus et les arguments des outils¶
Les modules nf-core prennent en charge deux types courants de remplacement de configuration : l'allocation des ressources (CPUs, mémoire, temps) et les arguments des outils via ext.args.
De nombreux outils en ligne de commande ont des arguments qui ne sont pas suffisamment courants pour être exposés comme paramètres du pipeline.
La convention ext.args vous permet de passer ces arguments à l'outil sous-jacent via un fichier de configuration.
Le fichier custom.config fourni dans votre répertoire de travail illustre ces deux types de remplacement :
| custom.config | |
|---|---|
Le premier bloc remplace l'allocation des ressources de FASTQC.
Par défaut, FASTQC utilise le label process_medium de base.config, qui alloue 6 CPUs et 36 Go de mémoire ; ici nous le limitons à 2 CPUs et 4 Go.
Le second bloc passe un argument supplémentaire à SEQTK_TRIM via ext.args.
Le flag -b 5 indique à seqtk trimfq de rogner 5 bases au début de chaque lecture en plus du rognage par qualité.
Exécutez le pipeline avec cette configuration :
Sortie de la commande
Le flag -c ajoute votre configuration par-dessus la configuration intégrée du pipeline.
Pour vérifier que le remplacement ext.args a bien pris effet, trouvez le hash du répertoire de travail de SEQTK_TRIM dans la sortie d'exécution (par exemple work/17/428668...) et vérifiez le fichier .command.sh à l'intérieur :
Sortie de la commande
Vous devriez voir -b 5 dans la commande seqtk trimfq.
Une chose importante à savoir concernant ext.args : si un module a déjà une valeur par défaut définie, votre valeur la remplacera complètement plutôt que de s'y ajouter.
Par exemple, le module FASTQC est configuré avec ext.args = '--quiet' par défaut (défini dans conf/modules.config) :
| conf/modules.config | |
|---|---|
Si vous définissez ext.args = '--kmers 8' pour FASTQC, le flag --quiet ne sera plus appliqué.
Pour conserver les deux, définissez ext.args = '--quiet --kmers 8'.
Vous devez toujours vérifier la configuration par défaut d'un module avant de remplacer ext.args.
À retenir¶
Vous savez comment obtenir de l'aide depuis un pipeline nf-core, définir des paramètres et comprendre comment ils sont validés, et personnaliser la configuration via des fichiers de configuration.
Et ensuite ?¶
Si vous souhaitez uniquement exécuter des pipelines nf-core, vous avez terminé !
Si vous souhaitez apprendre à développer vos propres pipelines selon les standards nf-core, faites une pause, puis passez à la Partie 2 lorsque vous vous sentez prêt·e. Vous apprendrez à créer votre propre pipeline compatible nf-core en utilisant les outils basés sur le template nf-core.